Running SQANTI3 rescue
Table of contents:
Introduction
As of SQANTI3 v5.1, a new module has been added to the SQANTI3 workflow for transcriptome characterization and quality control: SQANTI3 rescue.
Warning: SQANTI3 rescue has undergone significant changes as of release 6.0. The main changes have been in the logic of the rescue by mapping logic. In this wiki, only this new logic is explained. For information about the previous version of SQANTI3 rescue (v5.1 to v5.2.x), please refer to the archived wiki.
The SQANTI3 rescue algorithm is designed to be run after transcriptome filtering and uses the long read-based evidence provided by discarded isoforms (i.e. artifacts) to recover transcripts in the associated reference transcriptome. The idea behind this strategy is to avoid losing transcripts/genes that are detected as expressed by long read sequencing, but whose start/end/junctions could not be confidently validated using orthogonal data, resulting in the removal of those genes/transcripts from the transcriptome. More details about this can be found in the Motivation section below.
In particular, during the rescue, SQANTI3 will try to confidently assign each discarded artifact to the best matching reference transcript. As a result, SQANTI3 rescue will generate an expanded transcriptome GTF including a set of reference transcripts as well as the long read-defined isoforms that passed the filter. Optionally, requantification can be performed to reassign expression values to the rescued isoforms.
A new functionality of the rescue module is the requantification of the curated transcriptome. In this final step, the expression values of the transcripts that were not affected by filtering or rescue are kept intact, while the expression values of the rescued transcripts are transferred from their corresponding long read-defined artifact(s). More details about this can be found in the Requantification section below.
Motivation
The SQANTI3 rescue module was developed to mitigate the loss of biological diversity that occurs when stringent filtering removes long-read isoforms. Often, transcripts are discarded as "artifacts" because their start or end sites cannot be validated by orthogonal data, even though they contain high-confidence splice junctions that match the reference. By recovering these reference transcripts, the module ensures that legitimate genes and isoforms—detected by long-read sequencing but failing strict QC—are not permanently lost from the final curated transcriptome.
Beyond simple recovery, the module employs a rescue-by-mapping strategy to maintain a high-quality, non-redundant dataset. It matches discarded candidates (ISM, NIC, or NNC artifacts) to their best-matching reference targets and subjects them to the same quality filters used for the original transcriptome. This process also enables accurate requantification, allowing expression values from discarded artifacts to be transferred to rescued transcripts, ensuring that the final abundance levels reflect the true complexity of the sample.
SQANTI3 rescue strategy
Here is a summary of the SQ3 rescue workflow:
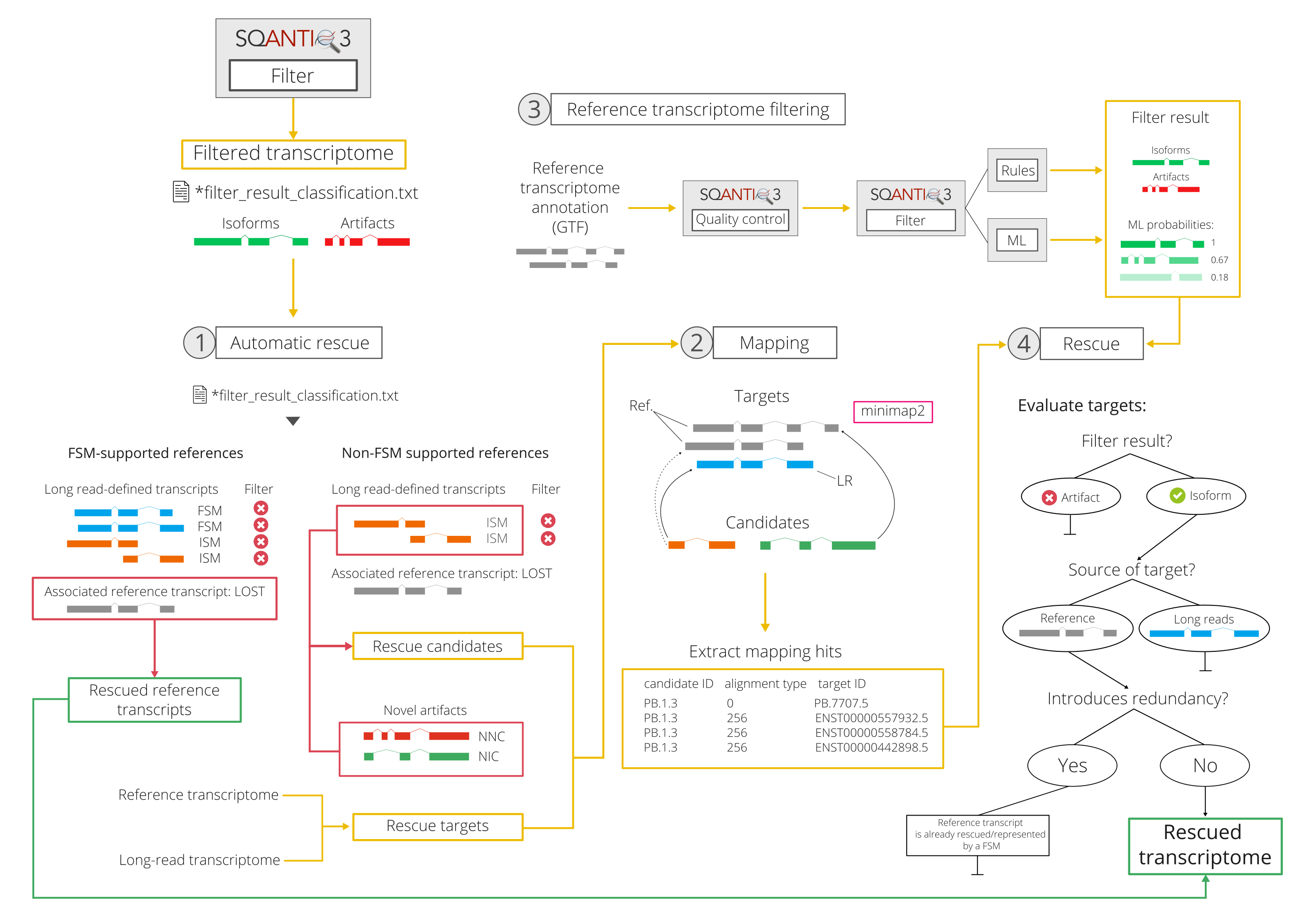
The SQANTI3 rescue algorithm consists in the following steps:
1.a Automatic rescue
As explained above, the rescue strategy in SQ3 was conceived to recover transcriptome diversity lost during filtering. This, among other things, means verifying that none of the reference-supported junction chains that were initially detected by long-reads are lost due to stringent artifact removal.
To achieve this during automatic rescue, all reference transcripts that were represented by at least one FSM in the original post-QC transcriptome are first retrieved -note that this information is available in the associated_transcript column of the *_classification.txt file. Then, those reference transcripts for which all FSM representatives were removed by the filter are rescued.
The previous analytic decision is justified because, in practice, any case where all FSMs with the same associated_transcript are removed can be interpreted as follows: 1) the TSS and/or TTS of the long read-defined transcript is different from that of the matching reference transcript, however, it could not be validated by SQ3 QC-supplied orthogonal data; and 2) the junctions are identical to those found in the reference, which can be interpreted as evidence that this isoform is real. As a result, SQ3 will not reintroduce any of the discarded FSMs, but the associated_transcript from the reference.
By default, SQANTI3 Rescue runs under --mode automatic. As a result, this is the only rescue step that SQANTI3 Rescue performs. The complete algorithm can be run by the --mode full argument.
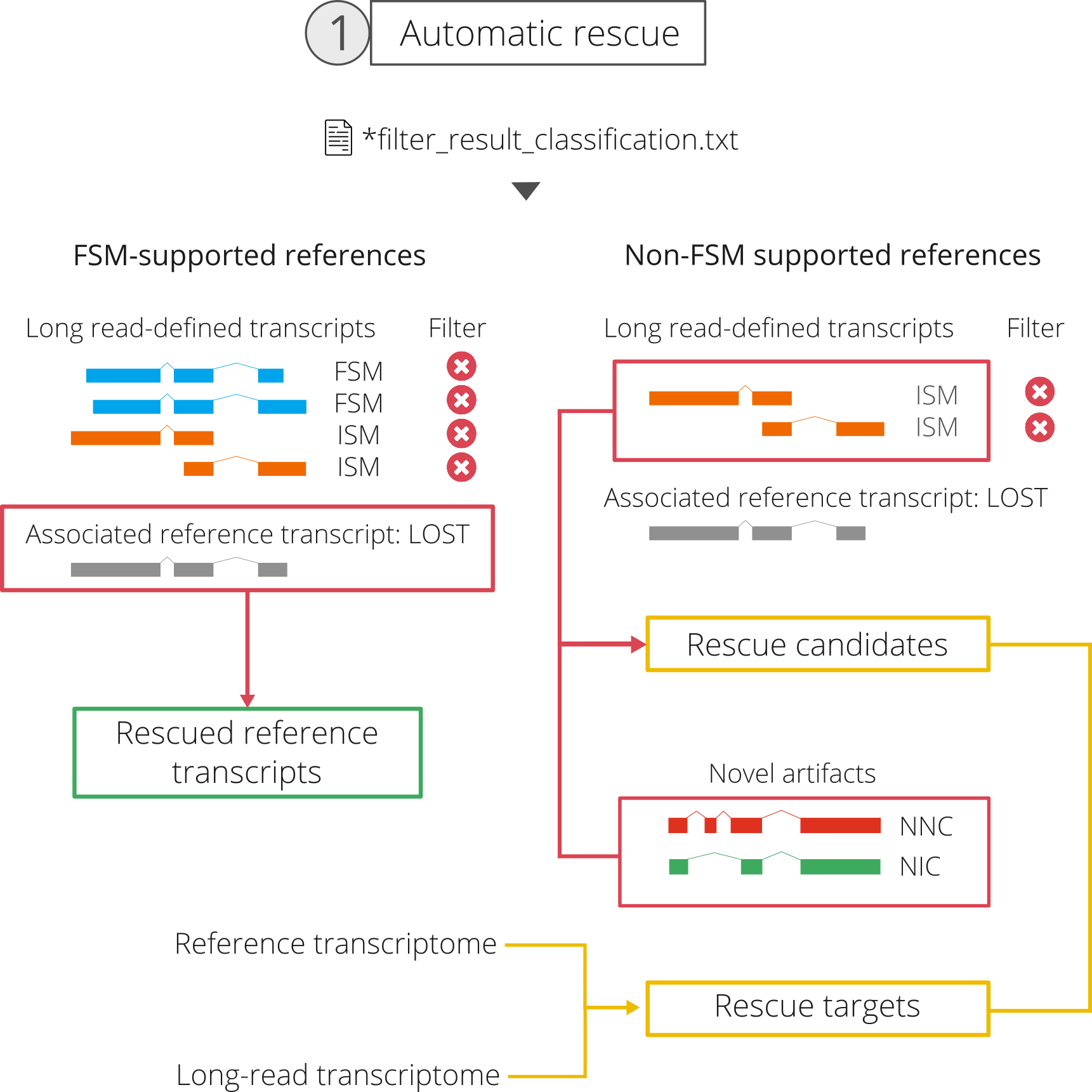
1.b Selection of rescue candidate and target transcripts
In spite of its potential to achieve the goals that we set for the rescue, the previous strategy does not consider ISM, NIC or NNC artifacts. These will be included in the rescue candidate group, i.e. transcripts classified as artifacts for which SQANTI3 will try to find a matching reference transcript to include in the final, curated transcriptome.
-
For ISM artifact transcripts, there are two possible situations:
-
There will be cases where the same discarded reference transcript is supported by FSM and ISM. ISM artifacts with an FSM artifact counterpart will therefore be "collapsed" into the rescued reference transcript during the automatic rescue step.
-
Conversely, there will be
associated_transcriptreferences that are only supported by one or more ISM. Those ISM artifacts that constituted evidence of a non-FSM supported reference will therefore be included in the rescue candidate list. -
For novel transcripts from the NIC and NNC categories, since there is no associated transcript information, all transcripts classified as artifacts will be included in the rescue candidate list.
As a result, we consider all reference or long read-defined transcripts from genes that have at least one rescue candidate to be rescue targets.
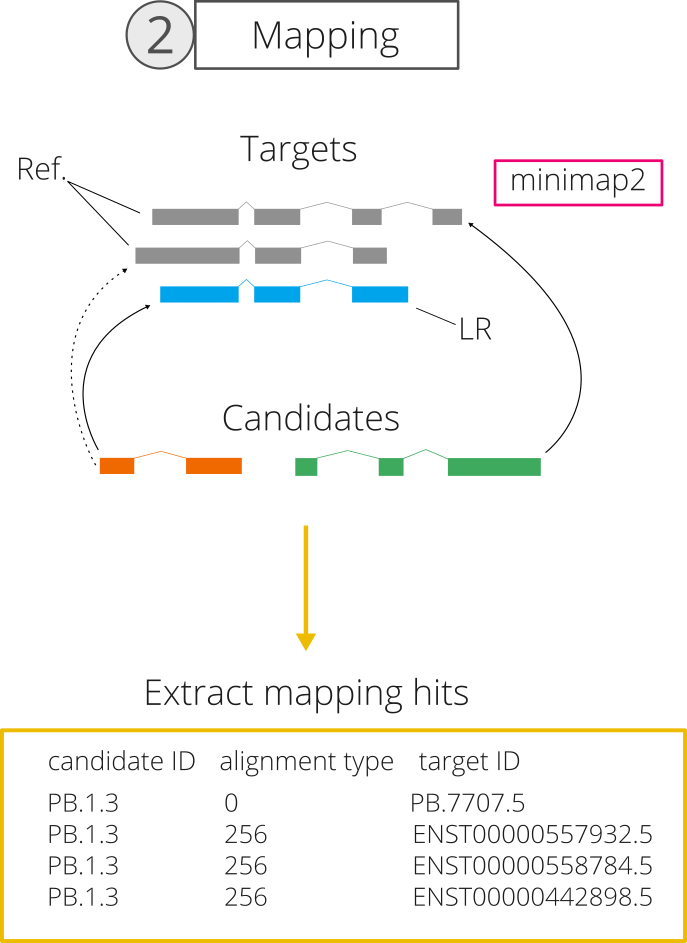
2. Mapping of candidates to targets
SQ3 rescue next tries to find matches between each rescue target and its same-gene candidates based on sequence similarity. To achieve this, we perform an internal mapping step using minimap2. In it, rescue candidates are considered to be "reads" and rescue targets are used as a "reference genome" in which each transcript sequence constitutes a different "chromosome".
To map candidates, we use the map-hifi option in minimap2 and the -a -x parameters:
minimap2 --secondary=yes -ax map-hifi rescue_targets.fasta rescue_candidates.fasta > mapped_rescue.sam
Finally, all candidate-target pairs obtained during mapping -referred to as mapping hits- are obtained from the output SAM file regardless of whether they are primary or secondary alignments. From the SAM file, we extract the alignment score of each hit, which is the number of match scores minus penalties for mismatches, gap opens and gap extensions (for more info, check minimap2 Github).
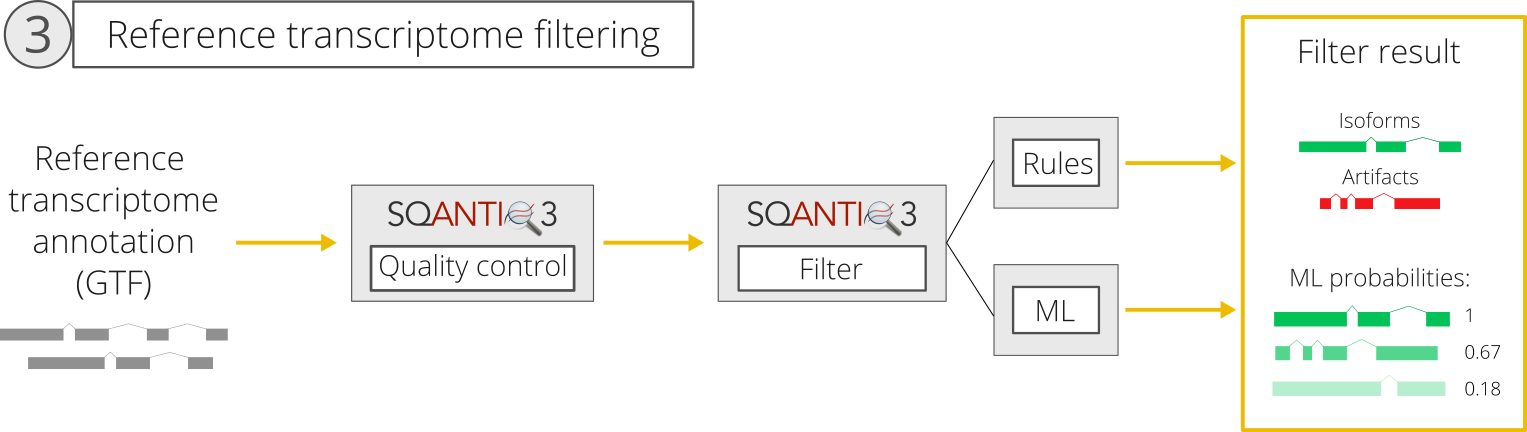
3. Application of SQ3 filter to the reference transcriptome
Validation of transcripts using orthogonal sources of data is an important part of the SQANTI3 philosophy. In consequence, the rescue strategy in SQ3 includes a validation step for all reference rescue targets before considering them for inclusion in the transcriptome, since no QC information is available for them (in contrast to long read-defined targets).
This requires users to run SQANTI3 quality control on the reference transcriptome and supply the output *_classification.txt file to SQANTI3 rescue using the --refClassif (-k) flag. This must be done using the same orthogonal data files that were used when running SQANTI3 QC for the long read-transcriptome, since the rescue is based on the assumption that the same evidence is required to validate all rescue targets. The only orthogona data that might be excluded is the count matrix, as it is specific to the long read-defined transcriptome.
Using the supplied reference classification file, SQ3 rescue will next apply SQ3 filter to the reference transcriptome. The filter to be applied will be specified by the rules or ml flags used when running the rescue. This means that, if you run SQ3 machine learning-based filter, you should also run the rescue using the ml option (and the same is true for the rules filter).
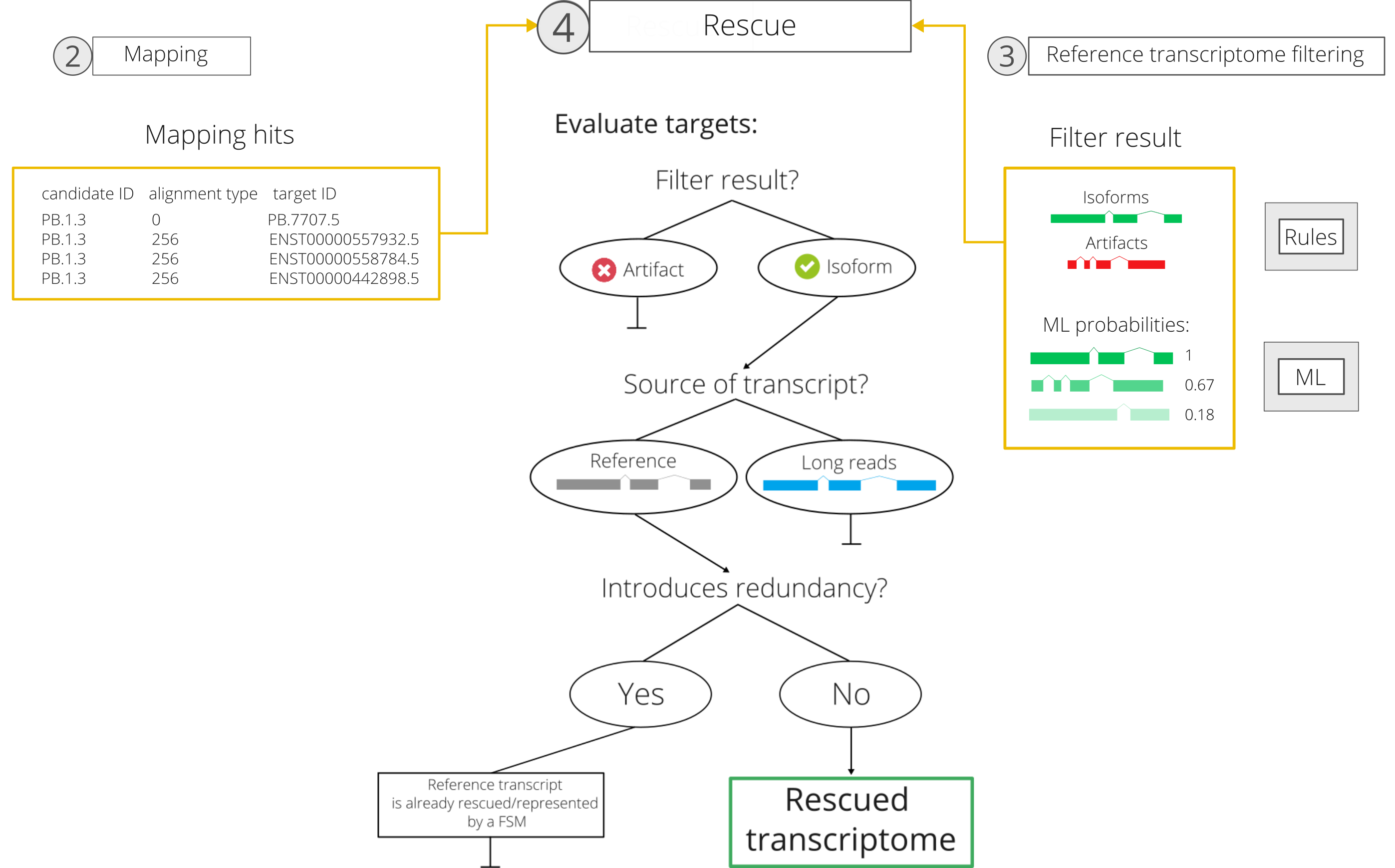
4. Rescue-by-mapping
In the final rescue step, hereby referred to as rescue-by-mapping, mapping hits (i.e. candidate-target pairs obtained during mapping) and reference transcriptome filter results are combined to generate a final list of reference transcripts to be rescued and reintroduced into the long read-defined transcriptome.
This part of the rescue can be divided into the following tasks/criteria:
-
SQ3 filtering of targets: first, candidate-target pairs are removed if the rescue target did not pass the filter (ML or rules). If the user is working with the rules filter, this will mean that the reference (or long-read) target transcript did not pass the rules defined in the JSON file. If the user is working with the ML filter, this will mean that the target transcript did not pass the specified ML probability threshold. In the case of the ML filter, however, only the target transcript(s) with maximum ML probability are selected to continue in the rescue.
-
Selection of the best match: for each rescue candidate, only the mapping hit with the highest score is retained. The score is obtained from multiplying the alignment score (obtained from minimap2) by the ML_POS_prob of the target transcript (if working with the ML filter) or by 1 (if working with the rules filter). In the case of ties, long-reads defined transcripts will be prioritized over reference transcripts as best matches. If there is still a tie, all of the tied targets will be retained as best matches.
-
Reintroduction of targets: finally, the best matching target transcripts that belong to the long-reads defined transcriptome are not reintroduced, since they are already present, to avoid redundancy. Conversely, the best matching target transcripts that belong to the reference transcriptome are added to the final list of rescued transcripts to be included in the curated transcriptome, if they were not already included during automatic rescue. In the case of a reference transcript being represented already by an FSM, it will still be reintroduced, as it is considered to be more similar to the long read-defined artifact than the FSM.
After performing this last filter of the rescue target list, SQ3 rescue outputs a list of rescued reference transcripts, which are then added to the long-read transcriptome GTF.
A new classification
5. Requantification
The requantification is applied to each transcript following one of the four scenarios:
1) A transcript was not impacted by either filtering or the rescue step; the expression level of such a transcript is intact.
2) A transcript was flagged by the filter as an artifact and was not rescued; the expression level is given to the transcript divergence of the associated gene (if any) or to the general transcript divergence pool.
3) A transcript was initially discarded and later rescued by the reference transcript; the initial expression level is transferred to the reference transcript.
4) One of the qualifying transcripts did not pass the redundancy test and was not included in the final rescue isoforms list, meaning that the transcript is already represented by a transcript model that was not affected by the filtering step; the expression levels from artifacts are summed up with the expression levels of the unaffected transcript.
Running SQ3 rescue
Similarly to the SQANTI3 filter, the SQANTI3 rescue is designed as a
dual implementation, depending on whether the rules or the machine learning filter was previously run. Therefore, the sqanti3_rescue.py
script requires a flag to be provided to activate either the ml or rules specific rescue.
usage: sqanti3_rescue.py [-h] {ml,rules} ...
Rescue artifacts discarded by the SQANTI3 filter, i.e. find closest match for
the artifacts in the reference transcriptome and add them to the
transcriptome.
positional arguments:
{ml,rules}
optional arguments:
-h, --help show this help message and exit
Input files and arguments
All in all, these are the arguments accepted by sqanti3_rescue.py rules:
usage: sqanti3_rescue.py [-h] --filter_class FILTER_CLASS -rg REFGTF -rf REFFASTA [--corrected_isoforms_fasta CORRECTED_ISOFORMS_FASTA] [--filtered_isoforms_gtf FILTERED_ISOFORMS_GTF] [-k REFCLASSIF] [--counts COUNTS]
[-e {all,fsm,none}] [--mode {automatic,full}] [-q] [-s {rules,ml}] [-j JSON_FILTER] [-r RANDOM_FOREST] [-t THRESHOLD] [-o OUTPUT] [-d DIR] [-c CPUS] [-v] [-l {ERROR,WARNING,INFO,DEBUG}]
Arguments explanation
Rescue artifacts discarded by the SQANTI3 filter, i.e. find closest match for the artifacts in the reference transcriptome and add them to the transcriptome. Choose between the filter applied: using rules or the
Machine-Learning approach.
options:
-h, --help show this help message and exit
Required arguments:
--filter_class FILTER_CLASS
SQANTI filter (ML or rules) output classification file.
-rg REFGTF, --refGTF REFGTF
Full path to reference transcriptome GTF used when running SQANTI3 QC.
-rf REFFASTA, --refFasta REFFASTA
Full path to reference genome FASTA used when running SQANTI3 QC.
Input options:
--corrected_isoforms_fasta CORRECTED_ISOFORMS_FASTA
FASTA file output by SQANTI3 QC (*_corrected.fasta), i.e. the full long read transcriptome.
--filtered_isoforms_gtf FILTERED_ISOFORMS_GTF
GTF file output by SQANTI3 filter (*.filtered.gtf).
-k REFCLASSIF, --refClassif REFCLASSIF
Full path to the classification file obtained when running SQANTI3 QC on the reference transcriptome. Mandatory when running the rescue on full mode
--counts COUNTS Isoforms abundance values: "Isoform" "Count". Column names may differ
Customization options:
-e {all,fsm,none}, --rescue_mono_exonic {all,fsm,none}
Whether or not to include mono-exonic artifacts in the rescue. Default: all
--mode {automatic,full}
If 'automatic' (default), only automatic rescue of FSM artifacts will be performed. If 'full', rescue will include mapping of ISM, NNC and NIC artifacts to find potential replacement isoforms.
-q, --requant Run requantification of the rescued isoforms.
-s {rules,ml}, --strategy {rules,ml}
Filter strategy used. Default: rules
Rules specific options:
-j JSON_FILTER, --json_filter JSON_FILTER
Full path to the JSON file including the rules used when running the SQANTI3 rules filter. Default: /home/pabloati/Programs/sqanti3/src/utilities/filter/filter_default.json
Machine Learning specific options:
-r RANDOM_FOREST, --random_forest RANDOM_FOREST
Full path to the randomforest.RData object obtained when running the SQANTI3 ML filter.
-t THRESHOLD, --threshold THRESHOLD
Machine learning probability threshold to filter eligible rescue targets (mapping hits). Default: 0.7
Output options:
-o OUTPUT, --output OUTPUT
Prefix for output files.
-d DIR, --dir DIR Directory for output files. Default: Directory where the script was run.
Extra options:
-c CPUS, --cpus CPUS Number of CPUs to use. Default: 4
-v, --version Display program version number.
-l {ERROR,WARNING,INFO,DEBUG}, --log_level {ERROR,WARNING,INFO,DEBUG}
Set the logging level INFO
Regardless of the rescue mode that is selected, SQ3 has the following common arguments, all of which are mandatory:
-
SQANTI filter classification: The SQANTI3 classification file of the isoforms after the filter step, which has the new columns that indicate which isoforms are artifacts and which are true isoforms.
-
Long read-defined isoforms: should be the FASTA file generated by SQANTI3 QC (
*_corrected.fasta). The isoform sequences will be used to extract rescue candidates and targets for mapping, and must be supplied via the--rescue_isoformsargument. -
Long read-defined transcriptome annotation (filtered): should be the GTF file output by SQANTI3 filter (
*.filtered.gtf). Rescued transcripts will be appended to this GTF file to generate the final curated transcriptome. This file must be supplied via the--rescue_gtfargument. -
Reference transcriptome annotation: reference GTF used to run SQANTI3 QC. This file will be used to extract rescue targets for mapping, and must be supplied via the
--refGTF(or-g) argument. -
Reference transcriptome classification file generated after running SQANTI3 QC on the reference transcriptome, which must be done previously to running the rescue and using the same orthogonal data as for long read-defined transcriptome QC. This file must be supplied via the
--refClassif(or-k) argument and will be used to evaluate reference rescue target support (see details above). -
Counts file containing expression values of transcript models before SQANTI3 filtering and rescue. This file is required if the
--requant(-q) module is used. It must include two columns: the first for the isoform ID and the second for the corresponding expression value. Column headers can vary. Provide this file using the--counts(or-c) argument; it will be used to re-evaluate the expression values of the filtered and rescued transcript models.
Additionally, the following parameters can be set to modify the behavior of the rescue algorithm:
-
Rescue mode: the
--modeargument can be used to extend the rescue algorithm to non-FSM artifacts by supplying--mode full. By default (--mode automatic), only the automatic rescue step is performed, enabling the rescue of high-confidence reference isoforms. -
Filter strategy: the user must specify which filter was used to filter the long read-defined transcriptome by providing either the
rulesormlflag when running the rescue. This will determine how reference rescue targets are evaluated during the rescue. -
Define rescue for mono-exon transcripts: the
-eflag (--rescue_mono_exonic) can be used to determine whether (and how) to perform the rescue on mono-exonic transcripts. The following options can be supplied: all(default): all mono-exon transcripts classified as artifacts will be considered as rescue candidates, regardless of the category that they are classified into.fsm: mono-exonic transcripts will only be considered for the rescue if they belong to the full-splice match (FSM) category. In this case, mono-exons will be rescued via the automatic rescue strategy.-
none: do not consider mono-exonic transcripts for the rescue (all mono-exons classified as artifacts will be discarded and no reference representatives included in the final transcriptome). -
Output directory and outfile prefix: the output directory can be set via the
-dflag (default: current directory). Output files will be named using the prefix provided using the-oargument (default: SQANTI3).
Rules arguments
In addition to the common arguments, the rules rescue requires the following specific files:
- Rules JSON file: using the
-jflag, the user must provide the JSON file used for running SQANTI3 rules filter on the long read-defined transcriptome. This same set of rules will be applied to the reference transcriptome to evaluate the reliability of reference rescue targets. Note that, to achieve this, SQANTI3 rescue requires the classification file output after running SQANTI3 QC on the reference to be provided via the--refClassifargument.
Machine learning rescue arguments
In addition to the common arguments, the machine learning rescue requires the following specific files:
-
Pre-trained random forest classifier: using the
-rflag, users must provide therandomforest.RDataobject that was obtained when running the machine learning filter on the long read-defined transcriptome. This classifier will be used to run the ML filter on reference transcriptome isoforms, i.e. obtain an ML probability value for reference rescue targets and evaluate their likelihood to be a true isoform based on orthogonal data supplied to SQANTI3 QC. Note that, to achieve this, SQANTI3 rescue requires the classification file output after running SQANTI3 QC on the reference to be provided via the--refClassifargument. -
ML probability threshold: minimum probability value that is required in order to consider a transcript to be a true isoform. It should be set using the
-jflag (default: 0.7). We recommend setting the same threshold that was used when running SQANTI3 filter.
Rescue output files
The final result of SQANTI3 rescue is a transcriptome GTF file and FASTA files - named *_rescued.gtf and *_rescued.fasta - including all the transcripts that were classified as isoforms by either the rules or the ML filter (already included in the supplied *prefix*.filtered.gtf file) and the rescued transcript isoforms that were reintroduced from the reference transcriptome (see rescue strategy section above).
Independently of running only rescue on automatic or full mode, two main files will be present:
*_rescue_inclusion_list.tsv: A single-column file including the IDs of all the reference transcripts that have been reintroduced into the transcriptome during the rescue.*_recue_table.tsv: A table summarizing the rescue results, with the same columns for both modes. All the columns are self-explanatory (origin refers to where the assigned transcript came from, i.e. reference or long read-defined transcriptome).
Mapping rescue specific files
In the case of the user running the rescue in full mode, additional output files will be generated, as explained below.
-
Rescue candidate files (for
--mode full): Briefly, rescue candidates are a group artifact transcripts from the ISM, NIC and NNC categories (which did not undergo automatic rescue) for which a reference transcript match needs to be found. Two rescue candidate files are generated: -
A rescue candidate ID list, named
*_rescue_candidates.tsv. -
A rescue candidate FASTA file named
*_rescue_candidates.fastacontaining the sequences of rescue candidates for mapping. -
Rescue target files (for
--mode full). As explained above, rescue targets are those reference and long read-defined transcripts for which there is a same-gene rescue candidate counterpart. During the rescue, candidates are mapped to targets using minimap2. Similarly to rescue candidates, two rescue target files are generated: -
ID list:
*_rescue_targets.tsv. -
FASTA file:
*_rescue_targets.fasta. -
Mapping results (for
--mode full): as a result of mapping with minimap2, two files are generated by SQ3 rescue: -
Minimap2 SAM file containing the mapping results, named
*_mapped_rescue.sam. -
A four-column table in which the mapping results are summarized, named
*_rescue_mapping_hits.tsv. This table has the following columns:- Column 1 (
rescue_candidate): rescue candidate ID. - Column 2 (
mapping_hit): ID of the rescue target that the candidate was successfully mapped to. - Column 3 (
alignment_type): SAM flag indicating the type of alignment. - Column 4 (
alignment_score): alignment score obtained from minimap2 for the candidate-target pair.
- Column 1 (
Rules-specific output
In addition to the common output files, when running --mode full, the rules rescue will create a new folder within the designated output directory to save rules filter output files, i.e. the results of running the rules filer on the reference transcriptome. This folder is named reference_rules_filter in all cases, while all files within it will have reference_ as prefix. The reference_RulesFilter_classification.txt file will be used by SQANTI3 rescue to get the rules filter result for reference transcripts.
Machine learning-specific output
In addition to the common output files listed above, when running --mode full, the ML rescue will also output a table including the result obtained after running the pre-trained ML classifier on the reference transcriptome. This file, named *_reference_isoform_predict.tsv, will contain reference transcript IDs in the first column and random forest classifier-generated probability values on the second. This probability can be interpreted as the extent to which the transcript is supported by SQANTI3 QC orthogonal data (short-reads, CAGE/polyA peaks, etc.). An example of this output is represented below:
isoform POS_MLprob
DQ459430 0.25
DQ516784 0.262
DQ516752 0.27
DQ668364 0.262
DQ883670 0.804
EF011062 0.512
DQ875385 0.758
Requantification output
If the --requant (-q) flag is used when running SQANTI3 rescue, two additional output files will be generated:
*_requantified_counts.tsv: a two-column table including the isoform IDs and their corresponding expression values after requantification.*_requantified_extended.tsv: an extended version of the previous table, with one extra column per sample with the counts of the isoform before requantification. In this table, the artifact isoforms are included.
Moreover, requantification will update the classification table FL columns, so the values reflect the new counts each isoform has after the whole pipeline. Note that the Transcript Divergence values will not be present in the classification table, only in the counts matrix.